

ECE2016 Eposter Presentations Endocrine tumours and neoplasia (68 abstracts)
Hyperparathyroidism-jaw tumour syndrome in Adolescence
Saba Hafeez , John Kalk , Aaisha Saqib & Louise Izzat
Medway Maritime hospital, Kent, UK.
Introduction: The hyperparathyroidism-jaw tumour syndrome is a rare autosomal, dominantly inherited disorder characterized by neoplastic or cystic lesions in parathyroid gland, jaws and the kidneys. With approximately 200 reported cases in literature this condition remains a challenge both diagnostically and in guiding further management.
Case report: Seventeen year old scholar was seen by the maxillofacial department at Medway Hospital with 2 painless swellings in his mouth, present for 2–3 months. Dental X-ray (OPG) showed resorption of bone at the sites of the swellings. Routine blood tests showed serum corrected calcium of 4.12 mmol/l (16.48 mg/dl), plasma PTH of 628 ng/l (10–65) and normal renal and thyroid functions. He had experienced significant polyuria and polydipsia for at least 2–3 years prior to this presentation. A CT scan of neck showed 2.5 cm × 2 cm right lower lobe parathyroid adenoma; ultrasound scan of his kidneys was normal. The tumour was resected: histology confirmed a benign parathyroid adenoma. There was no family history of any tumours.
Post-operatively the patient developed severe and prolonged ‘hungry bone syndrome’. He required prolonged HDU admission for intravenous calcium infusions despite high dose Alfacalcidol (4 mcg/day) and then oral calcium supplement at home. These are being reduced stepwise, at present as plasma PTH remains suppressed.
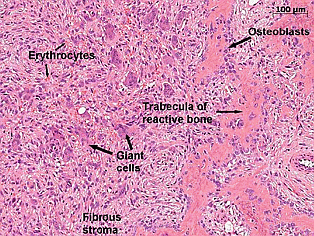
Soon after the parathyroidectomy the maxillary swellings were biopsied. The histology showed spindle cells, osteoclast giant cells and substantial bone formation.
Genetic tests showed the characteristic mutation in the CDC73 gene, confirming the diagnosis of the ‘hyperparathyroidism-jaw tumour syndrome’.
Discussion: On review of literature, in HPT-JT syndrome, jaw tumours are mostly composed of fibro cellular tissue without giant cells. In contrast our patient had multiple giant cell granulomas. In most cases, an affected person has one parent with the condition. This case illustrates consideration of genetic testing in younger patients presenting with primary hyperparathyroidism.
Article tools
My recent searches
My recently viewed abstracts

Endocrine Abstracts
ISSN 1470-3947 (print) | ISSN 1479-6848 (online)
© Bioscientifica 2024 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more