

EYES2019 7th ESE Young Endocrinologists and Scientists (EYES) Meeting Oral Presentations (67 abstracts)
Unusual cause of gigantism – Growth hormone releasing hormone (GHRH)-secreting pancreatic neuroendocrine tumour in a patient with multiple endocrine neoplasia type 1 (MEN1)
Vinaya Srirangam Nadhamuni 1 , Donato Iacovazzo 1 , Jane Evanson 2 , Jacqueline Trouillas 3 , Tom Kurzawinski 4 , Satya Bhattacharya 2 & Márta Korbonits 1
1Department of Endocrinology, William Harvey Research Institute, Bart’s and the London School of Medicine and Dentistry, Queen Mary, University of London, Mile End Rd, Bethnal Green, London, UK; 2St Bartholomew’s Hospital, W Smithfield, London, UK; 3Department of Pathology, Groupement Hospitalier Est, Hospices Civils de Lyon, Bron, France; 4Division of Endocrine Surgery, University College Hospital, Bloomsbury, London, UK.
Background: Gigantism is a rare condition with accelerated growth in childhood when the epiphyseal plates are not fused. Most cases are due to growth hormone (GH) secretion from a pituitary adenoma. Rarer causes of GH-related gigantism include somatotroph hyperplasia as part of McCune-Albright syndrome, Carney complex, X-linked acrogigantism or ectopic GHRH production.
Case presentation: An 18-year-old male with c.249_252delGTCT;p.I85Sfs MEN1 mutation, and history of insulinoma (age 10y) and primary hyperparathyroidism (age 15y) presented with accelerated childhood growth and gigantism. Height: 193.5 cm (+2.8S.D.S.).
Biochemistry: IGF1: 970 μg/l (>2× ULN), random GH: 39 μg/l, OGTT (GH nadir): 1.7 μg/l, GHRH: 327 ng/l (>5× ULN), calcitonin: 82 ng/l (>7× ULN), PRL: 213 mU/l (<329), PTH: 8.4 pmol/l (0.7–5.6), CgA: 44 pmol/l (<60).
Imaging: MRI abdomen: 40 mm (head) and 6 mm (tail) pancreatic tumours. MRI pituitary: bulky (9.9 mm craniocaudally). Review of previous imaging raised possibility of a 3 mm lesion in the right inferolateral anterior pituitary.
Treatment: Six-month 120 mg/month Lanreotide Autogel treatment reduced GHRH to 180 ng/l and IGF-1 to 778 μg/l. Whipple procedure was performed and 4 well-differentiated NETs were identified with the largest showing GHRH and calcitonin immunoreactivity with a ki-67 of 2%. Post-operatively, GHRH was undetectable and IGF-1 returned to normal. The 6 mm tail lesion was not resected and patient remains free of diabetes.
Conclusions: Childhood-onset GH excess in MEN1 syndrome patients is mostly due to GHRH-secreting pancreas NETs rather than pituitary adenoma. It is important to diagnose the underlying cause to prevent unnecessary pituitary surgery. Careful discussion between surgeon and patient is important regarding repeated pancreas surgeries in MEN1 syndrome.
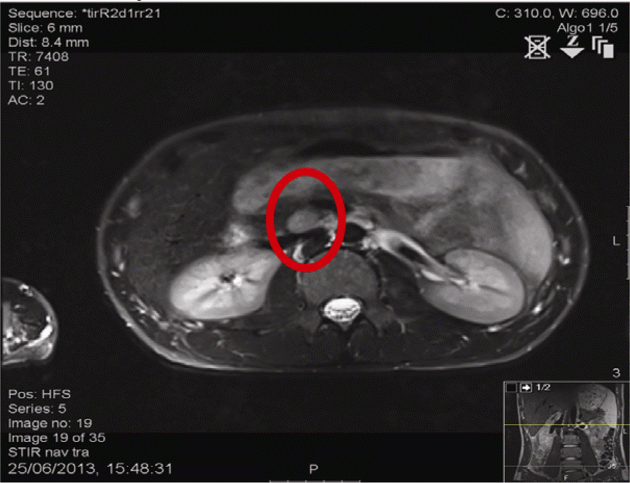
Figure 1 MRI abdomen: Circle highlights lesion in the head of the pancreas.
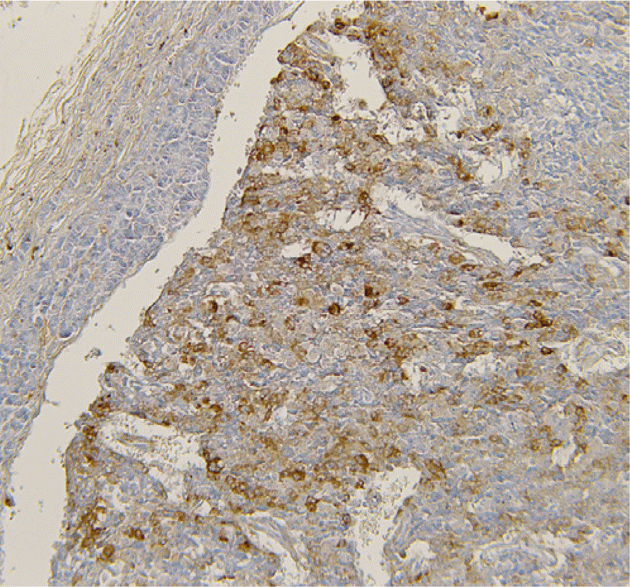
Figure 2 GHRH immunoreactivity in NET cells.
}
Article tools
My recent searches
My recently viewed abstracts
Authors

Endocrine Abstracts
ISSN 1470-3947 (print) | ISSN 1479-6848 (online)
© Bioscientifica 2026 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more