

BES2022 BES 2022 Abstracts (23 abstracts)
A rare cause of Cushing syndrome
Sol Bastiaan 1 , Carpriaux Marilyn 2 & De Leu Nico 1,3
1Department of Diabetology and Endocrinology, Universitair Ziekenhuis Brussel, UZ Brussel, (Vrije Universiteit Brussel, VUB), 1090, Brussels, Belgium; 2Department of Pathology, ASZ Aalst, 9300, Aalst, Belgium; 3Department of Endocrinology, ASZ Aalst, 9300, Aalst, Belgium
Introduction: Diagnosis of Cushing Syndrome (CS) is challenging due to its various non-specific symptoms, and multiple endogenous and exogenous causes. The incidence of endogenous CS is rare and estimated at 2 to 3 cases per million inhabitants per year in Europe (1). Primary bilateral macronodular adrenal hyperplasia (PBMAH) is an uncommon cause of endogenous ACTH-independent CS. It is a benign condition, characterized by the presence of bilateral macronodules (>1 cm), and autonomic hypersecretion of cortisol by the adrenal glands (2).
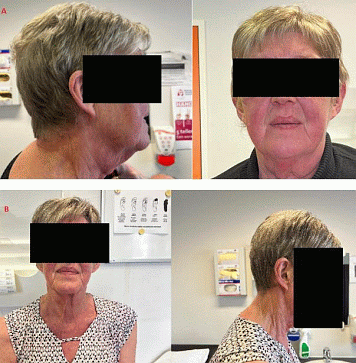
Case: A 64-year-old woman consulted our endocrinology clinic for persistent complaints of fatigue, depressed mood and reduced muscle strength. Her medical history consisted of chronic obstructive pulmonary disease, arterial hypertension, and paroxysmal atrial fibrillation. Physical examination showed a brittle skin with atrophy, and edema at the ankles. Proximal muscle wasting was present. The patient’s face was round, highly hyperemic with dorsocervical fat accumulation representing a typical “moon face” with “facial plethora and buffalo hump” (Figure 1A). 24h urine collection showed an elevated urinary-free cortisol of 305.5 μg/24h (normal reference:<120 mg/24h). Serum cortisol decreased insufficiently (30.6 μg/dL) after overnight 1 mg dexamethasone suppression (normal reference:<1.8 mg/dL). Morning blood analysis revealed a serum cortisol of 27.4 μg/dL (normal reference: 6,0 - 18,4 μg/dL), and a completely suppressed ACTH (<3.0 pg/mL) (normal reference: 7,20 - 63,30 pg/mL). Computed tomography (CT) and magnetic resonance imaging (MRI) of the adrenal glands showed bilateral hyperplastic adrenals sizing 58.9 by 29.7 mm on the right, and 55.6 by 42.6 mm on the left. With informed consent of the patient, bilateral adrenalectomy was performed with postoperative substitution of glucocorticoids (hydrocortisone 20 mg/day divided over 3 doses) and mineralocorticoids (fludrocortisone 100 μg once daily). Pathology report confirmed the diagnosis of PMBAH. At follow-up, a clear decrease of Cushing stigmata could be observed (Figure 1B). Genetic sequencing failed to reveal mutations in the ARMC5 gene.
Discussion: PMBAH is a rare cause of endogenous CS. Its exact prevalence is not established as its manifestation varies from subclinical, to severe and progressive forms (3). The pathophysiology of PBMAH is still under investigation. Although PBMAH is sporadic, ARMC5 germline mutations are described in up to 58% of patients, warranting pro-active analysis upon diagnosis (4). Although bilateral adrenalectomy with lifelong gluco- and mineralocorticoid substitution remains the best treatment option, unilateral adrenalectomy may be considered in specific cases (2). Currently, no additive value is gained from adrenal vein sampling- based cortisol lateralization ratios in the guidance of unilateral adrenalectomy (5).
Conclusion: In patients with suspicious signs such as atrophy of the skin with ecchymosis, red to purple-colored stretch marks, proximal muscle weakness and/or a plethoric/moon face, presence of CS should be considered. After confirmation of endogenous CS, differentiation between ACTH-dependent and ACTH-independent disease is essential. PMBAH is a rare cause of endogenous ACTH-independent CS. Although bilateral adrenalectomy remains the best curative option, unilateral resection can be considered if significant differences in adrenal size are present.
References: 1. Lindholm J et al. Incidence and late prognosis of Cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001; 86:117–123.
2. Vassiliadi DA et al. Diagnosis and management of primary bilateral macronodular adrenal hyperplasia. Endocr Relat Cancer. 2019 Oct 1;26(10).
3. Espiart S. et al. ARMC5 Mutations in a Large Cohort of Primary Macronodular Adrenal Hyperplasia: Clinical and Functional Consequences. J Clin Endocrinol Metab. 2015 Jun; 100(6): E926–E935.
4. Assié G et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med. 2013 Nov 28;369(22):2105–14.
5. Rubinstein G et al. The role of adrenal venous sampling (AVS) in primary bilateral macronodular adrenocortical hyperplasia (PBMAH): a study of 16 patients. Endocrine volume 76, pages 434–445 (2022).
 }
}
Article tools
My recent searches
My recently viewed abstracts

Endocrine Abstracts
ISSN 1470-3947 (print) | ISSN 1479-6848 (online)
© Bioscientifica 2026 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more