

BES2023 BES 2023 Section (29 abstracts)
Primary infertility: a rare presentation of adrenocortical carcinoma
Desmedt S , Van De Vijver A , Van Caenegem E & Vandewalle S
Background: Adrenocortical carcinomas (ACC) are rare and aggressive endocrine tumours with an annual incidence of 1 to 2 per million.(1) Frequently, patients present with signs of hormonal excess. The majority are cortisol excreting carcinomas, followed by aldosterone or androgen excess.(2) Estrogen secreting ACC are extremely rare. We present a case of estradiol and progesterone excess in primary infertility caused by an ACC.
Case presentation: A 32-year-old woman was referred to the endocrinology department because of persistently high oestradiol and progesterone levels after hormonal stimulation in the context of ICSI (intracytoplasmic sperm injection) due to male infertility. She had no relevant history besides resection of a cystadenoma at the right ovary 3 months before presentation and used 4 mg folic acid daily because of child wish and a family history of cleft lip. Patient had no complaints besides the impression of more pronounced hair at the upper lip and amenorrhea since hormonal stimulation. Sequential biochemical analysis revealed rapidly increasing levels of estradiol and progesterone under hormonal stimulation (recombinant FSH (follitropin alfa) 150 IU daily in a gonadotropin releasing hormone (GnRH) antagonist protocol with cetrorelix 0.25mg daily from day 6 of the stimulation) with persistent elevation of progesterone (see table 1) after cessation of hormonal therapy. Additional testing showed elevated DHEAS (972 µg/dl, ref. 25.9-460 µg/dl), 17-OH progesterone (12.0 ng/ml, ref. 0.35-4.13 ng/ml) and androstenedione (3.44 ng/ml, ref. 0.37-2.25 ng/ml) with mildly elevated testosterone (64.1 ng/dl, ref. 11-56 ng/dl). Estrone was extremely increased (2450 ng/l, ref. early follicular phase <150 ng/l). Inhibin B was low (36.7 ng/l, ref. <200 ng/l). Furthermore, morning cortisol was elevated at diagnosis (30.9 µg/dl). Low-dose dexamethasone (1 mg) did not suppress morning cortisol (31.3 µg/dl) and salivary cortisol at midnight was elevated (0.668 µg/dl). ACTH was suppressed in basal conditions (<1.51 pg/ml). Urinary normetanephrines and metanephrines were normal. CT scan without contrast enhancement showed an enormous mass of 30 HU, most likely situated at the left adrenal gland with an axial diameter of 12.9 cm (figure 1). Additional PET-CT demonstrated FDG-avidity and central necrosis in this lesion (figure 1) and no evidence of distant metastases. The tentative diagnosis of an estrogen and cortisol secreting ACC was made. After multidisciplinary evaluation the indication for maximal surgical resection was established and the patient underwent laparotomic left adrenalectomy in combination with left nephrectomy, splenectomy and resection of the pancreas tail. Solucortef treatment was started per-operatively. Anatomopathological evaluation confirmed the presence of ACC based on a Weiss score of 5 without vascular or lymph node invasion nor invasion in the adjacent structures. There was a low mitotic index of 5/50 HPF (high power field). After surgery, estrogen, estrone and progesterone levels soon dropped to baseline values and adrenal insufficiency occurred for which hydrocortisone substitution (20-10-10mg per day) was associated. Menstrual bleeding occurred 2 days postoperatively. Because of hypersecretion preoperatively and the tumour volume, treatment with mitotane was started 3 weeks postoperatively with acceptable tolerance. Patient was counselled that pregnancy is contraindicated until 1 year after cessation of treatment.(3) Re-evaluation with PET-CT is foreseen 3 months postoperatively. Discussion: Estrogen secreting ACC account for less than 2% of ACC. Feminizing adrenal tumours are a well-described but rare disease entity in males.(4) To our knowledge, this is the first case in literature of an estrogen and progesterone secreting ACC diagnosed because of primary infertility in a female patient.(5) Caution is needed in case of unexplained infertility or absence of hormonal variation during menstrual cycle in women.
| Start of stimulation | Day 13 of stimulation | Pre-operatively | 10 days postoperatively | Reference | |
| Estradiol | 55.9 | 5400 | 220.5 | <11.8 | 19-144 ng/l (follicular phase) |
| Progesterone | 1.14 | 2.35 | 3.34 | <0.21 | <1.4 µg/l (follicular phase) |
| Cortisol before 9 am | NA | NA | 30.9 | 3.3 | 5.3-22.4 µg/dl |
Conclusion: Although extremely rare, estrogen secreting ACC can present in premenopausal women with loss of menstrual cycle. Clinicians should be aware of this rare but severe disease entity.Table 1. Evolution of hormonal levels. NA: not availableFigure 1. CT-scan without contrast enhancement (left): giant mass of 13 cm at the left hypochondrium. PET-CT(right): FDG-avidity with central necrosis.
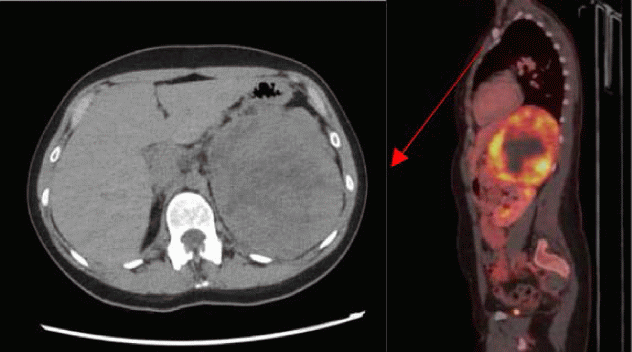
Figure 1. CT-scan without contrast enhancement (left): giant mass of 13 cm at the left hypochondrium. PET-CT(right): FDG-avidity with central necrosis.
References: 1. Ng L, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol. 2003;169(1):5. 2. Luton JP, Cerdas S, Billaud L, Thomas G, Guilhaume B, Bertagna X, Laudat MH, Louvel A, Chapuis Y, Blondeau P. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med. 1990;322(17):1195. 3. Puglisi, S., Basile, V., Sperone, P. et al. Pregnancy in patients with adrenocortical carcinoma: a case-based discussion. Rev Endocr Metab Disord 24, 85 96 (2023). 4. Chentli F, Beckie I, Azzoug S. Feminizing adrenocortical tumors: Literature review. Indian J Endocrinol Metab. 2015 May-Jun;19(3):332-9. 5. McKenna TJ, O’Connell Y, Cunningham S, McCabe M, Culliton M. Steroidogenesis in an estrogen-producing adrenal tumor in a young woman: comparison with steroid profiles associated with cortisol- and androgen-producing tumors. J Clin Endocrinol Metab. 1990 Jan;70(1):28-34.
}
Article tools
My recent searches
My recently viewed abstracts

Endocrine Abstracts
ISSN 1470-3947 (print) | ISSN 1479-6848 (online)
© Bioscientifica 2026 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more