

BES2021 Belgian Endocrine Society 2021 Abstracts (26 abstracts)
Unusual cause of neonatal salt wasting syndrome in a female with subsequent primary amenorrhoea
Verdickt Sébastien & Bex Marie
Department of Endocrinology, University Hospitals Leuven, Belgium
Introduction: Congenital adrenal hyperplasia (CAH) is the most frequent cause of primary adrenal insufficiency (PAI) in infants and children. The list of genetic causes of PAI has grown extensively in recent years(1). Associated features can provide a clue to identify the underlying defect.
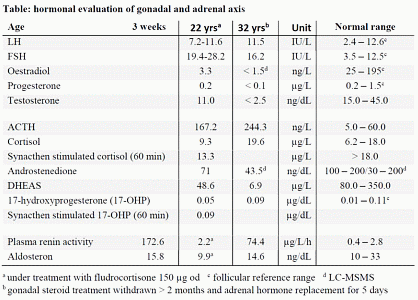
Case presentation: A 21-year-old female was referred for endocrinological advice about pregnancy. She had been diagnosed with isolated aldosteron deficiency at three weeks of age, presenting with failure to thrive, polyuria, renal salt loosing, hyponatraemia, hyperkalaemia, high plasma renin activity and low aldosteron. Cortisol was normal. During childhood and adolescence she was treated with fludrocortisone only. At age 14 she was diagnosed with late puberty. FSH was 15.1 U/L; LH 5.8 U/L and her karyotype 46,XX. Puberty was induced with ethinyloestradiol and was replaced later by oestroprogestogen contraception. Persistence of amenorrhea was confirmed at age 18, and birth control pills were resumed until her marriage 9 months before referral. A hormonal evaluation was performed in our clinic (table). Hypergonadotrophic hypogonadism and a partial cortisol deficiency with low adrenal androgens and moderately elevated ACTH were diagnosed. AMH was low. Hydrocortisone replacement therapy was added to fludrocortisone. She was referred to the fertility clinic and had one successful pregnancy using egg cell donation, giving birth to a daughter at age 25. Further attempts failed. In view of the combined deficiency of ovarian and adrenal steroids, including aldosterone, and a low 17-hydroxyprogesterone, a (partial) defect in the synthesis from cholesterol to pregnenolone was hypothesized. Genetic screening for mutations leading to lipoid CAH was however negative: sequencing of 7 exons of the STAR gene (8p11.2) and 9 exons of CYP11A1-(P450 cholesterol cleaving enzyme 15q23-24) gene was normal. At age 32 a urinary steroid profile was obtained after withdrawal of adrenal hormone replacement for some days and of gonadal steroids for two months. She developed limited salt wasting, with mild but symptomatic hypotension. Only androgen- and progesterone metabolites were low. Plasma renin activity and ACTH were elevated, with low normal aldosteron en normal cortisol (table), indicating partial deficiency of mineralocorticoids and glucocorticoids at most. Hypogonadism persisted with mildly elevated LH and FSH. As the gonadal dysfunction predominated a mutation in the steroidogenic factor-1 (SF-1, NR5A1) was suspected(2). A heterozygote missense variant was identified in exon 3 (c.104G> A, p.(Gly35Asp). This is located in the first zinc finger protein motif of the DNA binding domain. Her parents did not harbour the mutation, despite a history of premature menopause in the maternal family.
Discussion: Steroidogenic factor-1 (NR5A1) located on 9q33 is a nuclear receptor that regulates adrenal en reproductive development and function. It is also expressed in pituitary gonadotrophs, and the hypothalamus. Mostly heterozygous pathogenic variants are responsible for a wide spectrum of disorders of gonadal development. In 46,XY individuals phenotypes range from disorders of sex development (DSD) to oligo-azoospermia, and in 46, XX individuals from (familial) premature ovarian insufficiency to 46,XX ovotesticular DSD (p.R92 variants). Associated adrenal dysfunction is however rare less then 10 cases have been reported to date, mostly coupled to variants in p.G35 (heterozygous, like our patient) or in p.R92 (homozygous). SF-1 variants are an infrequent cause of unexplained childhood PAI, being identified in only 0.6% of 155 cases(3). Our case illustrates that the adrenal dysfunction does not progress with time. It therefore is unlikely to identify this variant if the PAI is diagnosed at older age.
References: 1. Primary adrenal insufficiency: new genetic causes and their long-term consequences; F. Buonocore & J.C. Achermann. Clinical Endocrinology 2020;92:11-20.
2. DAX-1 (Nr0B1) and steroidogenec factor-1 (SF-1 NR5A1) in human disease. J.P. Suntharalingham, F. Buonocore, A.J. Duncan & J. Achermann. Best Prac Res Clin Endocrinolo Metab 2015; 29:607-19.
3. Genetic analysis of pediatric primary adrenal insufficiency of unknown etiology: 25 years’ experience in the UK. F. Bunonocore et al. J Endocr. Soc 2021;5: bvab086
 }
}
Article tools
My recent searches
My recently viewed abstracts

Endocrine Abstracts
ISSN 1470-3947 (print) | ISSN 1479-6848 (online)
© Bioscientifica 2026 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more